
1. INTRODUCTION
To ensure the appropriate utilization of the designated species and to obtain important biological species information relevant to the restoration of cultural properties, archaeology, and forensic science, the accurate identification of wood species is important (Dumolin-Lapegue et al., 1999; Deguilloux et al., 2003; Rachmayanti et al., 2009). The identification of wood species is recognized as an important method for solving cases of illegal timber logging to protect forests (Rachmayanti et al., 2006; Dormontt et al., 2015). Most wood species have been identified successfully by anatomical observations using a microscope (Eom and Park, 2018; Kim and Choi, 2016; Kwon et al., 2017). However, the microscopic identification of wood species requires trained and experienced professionals to compare the anatomical features of different wood species (Wheeler et al., 1989; Wheeler and Baas, 1998; Ogata et al., 2008; Rachmayanti et al., 2009). In addition, the methods of anatomical identification of wood have limitations when specimens are structurally similar to each other, such as closely related species or subspecies (Marco et al., 1994; Feuillat et al., 1997; Gasson, 2011; Jiao et al., 2015). To overcome these drawbacks, DNA sequencing is used as an alternative method of wood identification (Dumolin-Lapegue et al., 1999; Jiao et al., 2015). DNA sequencing enables the identification of wood at the molecular level and facilitates distinction between closely related species (Hebert et al., 2003; Hardy et al., 2006; Linacre and Tobe, 2011; Degen et al., 2013). Living plant cells actively use and maintain DNA; therefore, it is easy to extract and sequence DNA from living cells. In contrast, wood cells die after harvesting, resulting in fragmentation of DNA (Bär et al., 1988; Lindahl, 1993; Cano, 1996; Deguilloux et al., 2002; Pääbo et al., 2004; Rachmayanti et al., 2009). Therefore, it is difficult to extract DNA from wood. In addition, the methods used to extract DNA from living plant cells cannot be applied to wood because wood is subjected to a drying process to maintain its quality. For these reasons, the molecular identification of wood species has not been widely used, although it provides more precise information. These limitations hamper the establishment of a DNA database for the identification of wood species further.
To identify wood species using DNA sequencing, appropriate DNA markers must be selected. An ideal DNA marker is easily amplified from the fragmented DNA in wood samples and is able to simultaneously distinguish the samples. (Budowle and van Daal, 2008; Finkeldey et al., 2010). To meet these criteria, the chloroplast genome, existing in multiple copies in a single plant cell, is used for species identification (Deguilloux et al., 2002, 2003; Gailing et al., 2003; Indrioko et al., 2006). Single nucleotide polymorphisms (SNPs) in rpoB have been proposed for species identification in plants (Al-Qurainy et al., 2011; Khan et al., 2012). In this study, our purpose was to develop and validate a pretreatment method for extracting DNA from dried wood by hydration using water and pulverization using sandpaper. No special pretreatment method for extracting DNA from wood has been proposed previously. Based on our pretreatment method, we identified dried pitch pine using the extracted chloroplast gene rpoB, which encodes the β-subunit of RNA polymerase (National Center for Biotechnology Information accession number: JN854163.1), as a DNA marker. It is difficult to introduce random mutations in the conserved region because of its biological function; however, SNPs can be observed at nonconserved regions.
2. MATERIALS and METHODS
Logs of sapwood (5 cm × 2.5 cm × 1.5 cm) of Pinus rigida were harvested in 2014. Six types of sandpaper (Chunil Grinding Co., Ltd., Seoul, Korea) with different roughness (40, 50, 60, 80, 100, and 220 grit) were used.
The wood specimens were autoclaved at 121°C for 20 min to eliminate any contamination. To remove surface contaminants further, a layer of approximately 1-mm thickness was removed from the surface of the wood specimens using sterilized sandpaper. Sandpaper with varying degrees of roughness was used to pulverize the specimens, and an optical microscope (Axio Imager.A1, Carl Zeiss Vision Korea Co., Ltd., Seoul, Korea) was used to observe the particle size of the powder. The wood powder (20 mg) was collected in a sterilized centrifuge tube, and 200 μL of sterilized distilled water was added as a hydration solvent. To suppress microbial growth, the mixtures of wood powder and water were incubated at 4°C.
DNA was extracted from the hydrated wood powder with a DNeasy Blood & Tissue Kit (catalog number: 69504; Qiagen Korea, Ltd., Seoul, Korea) according to the manufacturer's instructions. Briefly, 600 μL of AP1 buffer and 6 μL of RNase A (100 mg/mL) were added to the hydrated wood powder and mixed thoroughly. The sample was incubated at 65°C for 10 min and was inverted every 2 min for mixing. After incubation, 260 μL of P3 buffer was added, and the sample was mixed well by inverting the tube. The fully mixed sample was placed in ice for 5 min and was centrifuged at 13,500 rpm for 10 min. The supernatant was removed using a pipette tip with a truncated end and was transferred to a QIAshredder Mini Spin Column (Qiagen Korea, Ltd.). After centrifugation at 13,500 rpm for 2 min, the solution was added to a new centrifuge tube. For each sample, a 1.5-fold volume of AW1 buffer was added to the solution, and the sample was immediately mixed using a pipette tip with a truncated end. The mixture (650 μL) was added to the DNeasy Mini Spin Column (Qiagen Korea, Ltd.) and was centrifuged at 8,000 rpm for 1 min. The flow-through solution was discarded, and the remaining mixture was added to the same column and was centrifuged at 8,000 rpm for 1 min. The column was placed in a new collection tube, and 500 μL of AW2 buffer was added to the column and was centrifuged at 8,000 rpm for 1 min. After the flow-through solution was discarded, the collection tube was remounted, and 500 μL of AW2 buffer was added again and centrifuged at 13,500 rpm for 2 min. The flow-through solution and the collection tube were discarded. The column was moved to a new centrifuge tube and was covered with clean tissue paper (KIMTECH, YuHan-Kimberly, Ltd., Seoul, Korea). The column was dried at room temperature for 40 min. Subsequently, 50 μL of AE buffer was placed in the center of the column, was incubated at room temperature for 5 min, and then was centrifuged at 8,000 rpm for 1 min. The DNA suspension obtained was stored in a freezer at ‒20°C.
A 174-bp fragment of the rpoB gene was amplified by PCR using the primers RPOB-1F (5-GCTTACACGA GCCCATATCC-3) and RPOB-1R (5-GGGATTT ACAGAATCGTGGTG-3) (Sun and Feng, 2011). PCR was performed in a 20-μL volume containing 2 μL of 10X Taq reaction buffer, 0.4 μL of 10 mM dNTP mixture, 0.8 μL of each 10 pM primer, 0.1 μL of BioFACT™ Taq DNA polymerase (5 U/μL; BIOFACT Co., Ltd., Daejeon, Korea), 2 μL of extracted DNA template, and 13.9 μL of water using GenePro Thermal Cycler (TC-E-48D; Hangzhou Bioer Technology Co., Ltd., Hangzhou, China). The PCR conditions were as follows: initial denaturation at 94°C for 5 min, followed by 40 cycles of denaturation at 94°C for 1 min, annealing at 55°C for 1 min, and extension at 72°C for 30 s, and a final extension at 72°C for 5 min.
Two microliters of the rpoB gene amplified by PCR were separated by gel electrophoresis using 1.5% agarose gel. For gel extraction, buffer from the QIAquick Gel Extraction Kit (catalog number: 28706; Qiagen Korea, Ltd.) and columns from the HiGene™ Gel & PCR Purification System (catalog number: GP104-100; BIOFACT Co., Ltd.) were used, and the DNA sample was extracted from the gel according to the instructions of the QIAquick Gel Extraction Kit. The purified DNA was confirmed by gel electrophoresis using 1.5% agarose gel.
Purified PCR products were bidirectionally sequenced by BIOFACT Co., Ltd. A 133-nucleotide sequence was obtained (excluding the primer sequences) and was used as a query to search the nucleotide database of the National Center for Biotechnology Information (NCBI; https://www.ncbi.nlm.nih.gov/) using the nucleotide BLAST algorithm. The COBALT program available at the NCBI website was used to align all the similar sequences.
3. RESULTS and DISCUSSION
To extract DNA from a biological sample, cells must be ruptured. Living plant cells are surrounded by cell walls and membranes, which must be broken by enzymatic and physical methods. Dried wood is sturdy enough to withstand physical treatment. Therefore, methods used to break living plant cells are not applicable to wood. In this study, we used sandpaper with various degrees of roughness to pulverize the wood and break open the cells.
A microscope was used to observe the particle size of the wood powder obtained from sandpaper treatment (Fig. 1). The particle size of the wood powder decreased with increase in the grit number, a measure of the roughness of the sandpaper. With 60-grit sandpaper, wood particles of around 100 μm in diameter were obtained. Because plant cells vary in size from 10 to 100 μm (Smith, 2017), our results indicate that sandpaper with grit numbers of 60 or higher can break cells in wood samples. The rpoB gene was successfully amplified six times from template DNA isolated from wood powder that was obtained using 60-grit sandpaper and was hydrated for 2 to 3 days.

This sandpaper method does not require special material or equipment, except for sandpaper, which can be easily purchased at low cost. In addition, because sandpaper is cheap, it can be used once for each sample, which prevents contamination of samples and facilitates the treatment of numerous samples in a relatively short time. Using sandpaper for pulverizing wood does not require special skills.
DNA in the wood is fragmented and partially degraded and possibly sticks to the internal structure of the cell during the drying process (Rachmayanti et al., 2009). Even if the cell's structural integrity is destroyed by the sandpaper, the attached DNA cannot be eluted by a general DNA extraction method. In this study, we used a hydration process to elute the attached DNA. We determined the hydration time that was most effective for isolating DNA from wood powder.
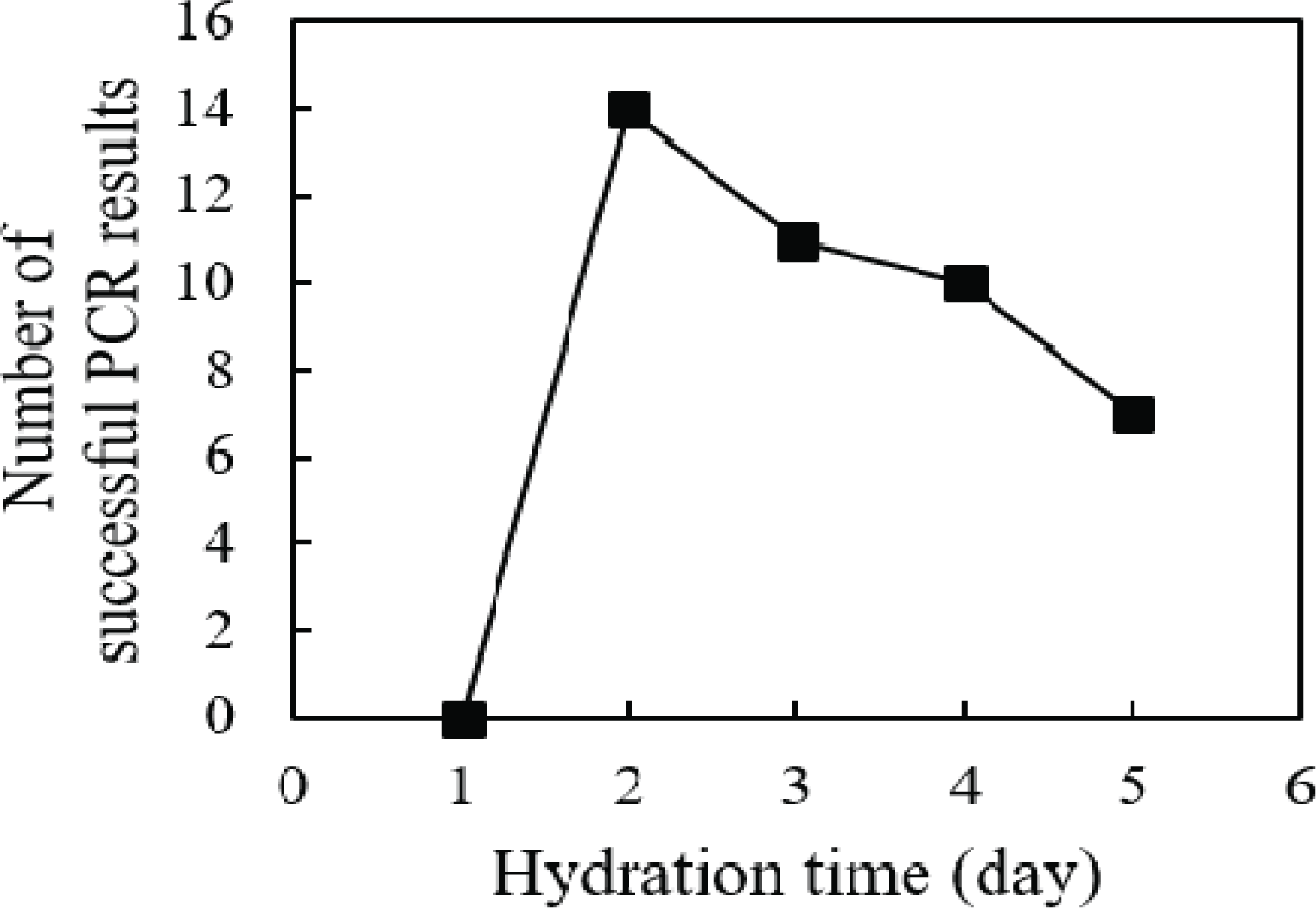
To isolate DNA, wood powder obtained with the use of sandpaper was hydrated with distilled water for 1 to 5 days. The rpoB gene was amplified from the DNA isolated from hydrated wood powder using PCR (Fig. 2). No amplification was obtained from DNA samples hydrated for 1 day, regardless of the roughness of the sandpaper. Amplification of the rpoB gene was successfully observed in samples hydrated for 2 days or more. However, when the hydration period was 4 days or longer, the amplification success rate decreased. This observation supports the hypothesis of this study that the DNA in wood cells attaches to cell structures during the drying process. The decrease in the PCR success rate with prolonged hydration suggests that the eluted DNA may be degraded by contaminated enzymes or microorganisms during the long incubation period, even when the wood powder is hydrated at 4°C. Overall, our data indicate that the optimal hydration time of wood powder for DNA elution is 2 days; this hydration time minimizes the degradation of eluted DNA while obtaining a sufficient DNA yield.
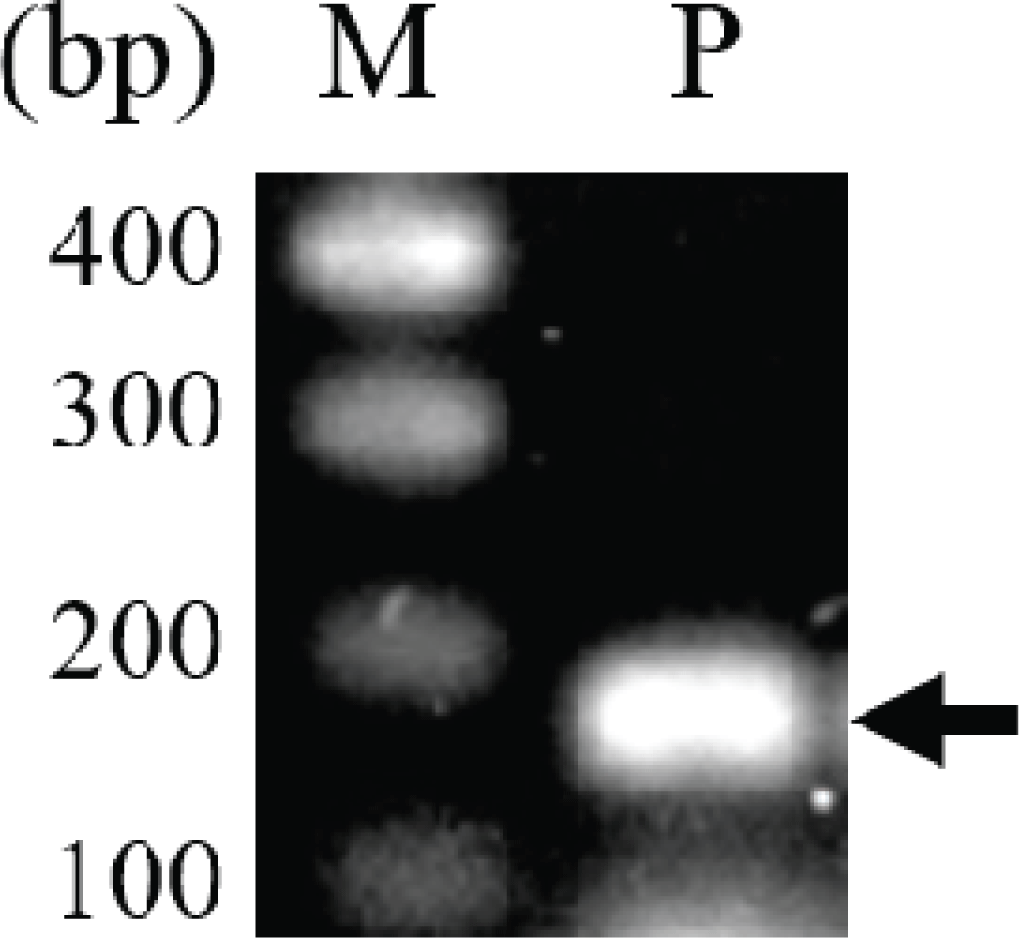
In previous studies, intergenic spacer DNA sequences, psbA-trnH (Hong et al., 2014), atpF-atpH (Hong et al., 2014), and trnT-trnL (Um et al., 2014), in the chloroplast were used for taxonomic studies of the genus Pinus. In this study, SNPs of the rpoB gene, which are used in classification of many plants, including the genus Pinus (Al-Qurainy et al., 2011; Khan et al., 2012), were analyzed for the evaluation of both the usefulness of rpoB for identification of P. rigida and the efficiency of the pulverization and hydration pretreatment for DNA extraction. A 174-bp fragment of the rpoB gene encoding the β-subunit of the chloroplast RNA polymerase was amplified (Fig. 3). The length of the PCR product was 174 bp including the primer sequences and 133 bp excluding the primer sequences (Fig. 4 and Supplementary Fig. 1). The nucleotide sequence of the amplified rpoB gene was used to search for similar sequences in the Pinus genus in the NCBI nucleotide database. Sequence analysis revealed SNPs at six locations in the gene sequence, and five groups were observed (Supplementary Fig. 1). The nucleotide sequences of representative genes of each group were aligned with those of rpoB obtained in this study (Fig. 4). The nucleotide sequence of rpoB in P. rigida was identical to that of the group containing P. koraiensis rpoB (NCBI accession number: AY228468). Five of the six SNPs differed from P. rigidarpoB (NCBI accession number: JN854163), which was reported previously in the NCBI nucleotide database as belonging to the second group. These results suggest that SNPs in the rpoB gene, as identified previously (Sun and Feng, 2011) and analyzed in this study, may not be suitable for species identification; however, these SNPs can be used to determine the origin of different wood subspecies or individual samples of wood.

4. CONCLUSION
In this study, we propose a pretreatment method for wood samples that involves pulverizing the wood samples using 60-grit sandpaper followed by hydration with water for 2 days for DNA extraction. Pulverization of wood using sandpaper is inexpensive, requires no special equipment or skills, and eliminates the chance of contamination. DNA isolated by this method was a good template to amplify the rpoB gene. Sequence analysis revealed five groups of SNPs in the rpoB gene in the genus Pinus. Although these SNPs were not suitable for species identification, they can potentially be used to determine the origin of different wood subspecies or individual samples of wood.