






1. INTRODUCTION
Amino plastic resins such as urea-formaldehyde (UF) resins, melamine-urea-formaldehyde (MUF) resins, and melamine-formaldehyde (MF) resins are thermosetting amino resins. These amino resins had been used as adhesives for the manufacture of wood-based composites and studied by many researchers (Akinterinwa et al., 2020; Dunky, 1998; He et al., 2012; Jeong et al., 2020; Jeong and Park, 2019; Kim and Park, 2021; Lubis et al., 2019a, 2019b; Lubis and Park, 2020; Moslemi et al., 2020; Myers et al., 1986; Myers, 1984; Park, 2007; Park and Kim, 2007; Park et al., 2017, 2006; Park and Causin, 2013; Park and Jeong, 2011; Pizzi and Valenzuela, 1994; Stuligross and Koutsky, 1985; Veigel et al., 2012; Wibowo et al., 2020a, 2020b; Wibowo and Park, 2020a, 2020b). In particular, UF resins produced by reacting urea with formaldehyde as raw materials are most commonly used adhesives for wood-based composite panels such as plywood, particleboard, medium density fiberboard (Park et al., 2006) because of their high reactivity, short curing time, low cost, colorless, and good bonding strength (Dunky, 1998). In fact, the production of formaldehyde-based resins in Republic of Korea in 2018 accounted for 32.5% (about 228,000 tons) of total resins, of which UF resins accounts for about 60% (about 138,000 tons). However, they have a problem of formaldehyde emission, which is particularly serious for interior applications (He et al., 2012; Park and Jeong, 2011).
In general, it is well known that the formaldehyde emission is due to either the hydrolysis of cured UF resins or free-formaldehyde (Myers et al., 1986). Thus, a lot of research was conducted to decrease the formaldehyde emission. Up to now, one of the widely accepted methods is to have low formaldehyde-to-urea (F/U) molar ratio for the synthesis of UF resins (Myers, 1984; Park et al., 2006; Pizzi and Valenzuela, 1994). But, low-molar-ratio (LMR) UF resins have weaknesses such as low reactivity, long gelation time, and poor adhesion (Wibowo et al., 2020a). Park (2007) reported that lowering of F/U molar ratio made gel time increased and viscosity decreased. Park and Kim (2007) explained why LMR UF reins showed poor adhesion performance using DMA. Park et al. (2006) reported that decreasing of F/U molar ratio not only decreased thermal curing of UF resins but also decreased the internal bond strength (IB) of particleboard. These weaknesses of LMR UF resins are partially due to the crystallinity of LMR UF resins (Singh et al., 2014).
The crystallinity of UF resins already reported by many authors. Stuligross and Koutsky (1985) reported that UF resins showed colloidal properties and crystallinity with lowering F/U molar ratio, whose crystal structure did not change but the crystallinity was changed by formulation. Singh et al. (2014) found the spherical structures in cured LMR UF resins via microscopy. Park and Jeong (2011) reported that hardener type and addition level, curing time and temperature did not affect to the crystalline regions or peak position of LMR UF resins. Park and Causin (2013) reported that the crystallinity and crystallite size of cured UF resins increased with F/U molar ratio, and the average distance between domains decreased with F/U molar ratio. In recent years, Wibowo and Park (2020b) calculated the crystallinity of cured LMR UF resins with deconvolution method with Gaussian function, and found that the addition level of hardener was greater contributor to the crystallinity than hardener type, curing temperature, and curing time. Wibowo et al. (2020b) reported that the crystalline structure in LMR UF resins was formed by hydrogen bonds between linear molecules, resulting from oxymethylene and branched methylene ether linkage cleavages, and they also mentioned that the crystalline domain in LMR UF resins was formed at the addition reaction of the final stage of resin synthesis. In summary, the crystallinity of LMR UF resins resulted from linear molecules in the crystalline domain and these harmed the adhesion of LMR UF resins because the crystalline domain hindered the formation of cross-linked structures. So, it is necessary to convert the crystalline structure of LMR UF resins to amorphous structures to improve the adhesion. In recent, there were a few reports on the crystallinity of LMR UF resins. Wibowo et al. (2020a) modified the crystalline structure of LMR UF resins to amorphous structure with modified nanoclay by preventing the formation of hydrogen bonds via the intercalation. Wibowo and Park (2020a) used multi-reactive melamine to decrease the crystallinity of low- molar-ratio UF resins by preventing the formation of hydrogen bonds between linear molecules of LMR UF resins.
Cellulose nanomaterials (CNMs) have at least one nanoscale dimension (1-100 nm), which not only exhibit long, flexible, entangled networks but also have abundant hydroxyl groups (Nechyporchuk et al., 2016; Phanthong et al., 2018). Due to these properties, CNMs are expected to prevent the formation of hydrogen bonds between linear molecules of LMR UF resins. Thus, in this study, CNMs such as cellulose microfibrils (CMFs), cellulose nanofibrils (CNFs), TEMPO- oxidized nanocellulose (TEMPO-CNFs) were used to modify the crystalline domain of LMR UF resins. Furthermore, two modification methods of either in situ modification or post-mixing were employed in this work to find a better method of modifying LMR UF resins. In addition, we used various characterization techniques such as attenuated total reflection (ATR-IR) spectroscopy, transmission electron microscope (TEM), X-ray diffraction (XRD) for the modified LMR UF resins with CNMs.
2. MATERIALS and METHODS
Technical-grade urea (99%), formalin (37 wt%, Daejung Chemical, Korea), and aqueous solutions of formic acid (20 wt%) and NaOH (20 wt%) were used to synthesis the low-molar-ratio UF resins. NH4Cl (20 wt%) was used as hardeners. Commercial CMFs (1 wt%) and CNFs (1 wt%) were purchased from CNNT Co. Ltd, Suwon, Korea, and used for the modification of LMR UF resins. It is reported that these CNMs prepared by the ACC had similar crystallinity, except the component ratio of cellulose Iα and Iβ (Kose et al., 2011). TEMPO-CNFs (2 wt%) were received from Moorim Paper, Ulsan, Korea, and used for the modification of LMR UF resins. It is also reported that the TEMPO-CNFs are maintaining their crystallinity (Isogai et al., 2011).
Attenuated total reflection (ATR)-infrared (IR) spectroscopy (ALPHA-P model, Bruker Optics GmbH Ettlingen, Germany) equipped with an ATR universal accessory were used with IR spectroscopy to characterize the functional groups of CMFs, CNFs and TEMPO-CNFs. All CNMs were oven-dried at 105 ℃ for 24 h, and then scanned for 32 times in the range of 400-4000 cm-1 at a 4 cm-1 resolution for the min- max normalization of the spectrum of each sample.
The morphology and dimensions of the CMFs, CNFs, and TEMPO-CNFs were characterized with a TEM (JEM-2100 F, JEOL, Japan), using the reported procedures (Khanjanzadeh and Park, 2020). Each of these samples was diluted to a concentration of 0.01 wt%. A droplet of this suspension was dropped on a copper grid and dried at room temperature. Subsequently, the specimen was stained by putting a droplet of 3% uranyl acetate solution on the grid to intensify the contradistinction of images. The excess stain solution was absorbed with filter paper and dried at room temperature. The dimensions of the CMFs, CNFs, and TEMPO- CNFs were measured with the Image-processing software (IMT solution ver.22.5, British Columbia, Canada).
Neat UF resins used for this study were synthesized by two-step alkaline-acid process with F/U molar ratio of 1.0 (Singh et al., 2014). Formalin solution was added into the four-neck reactor and pH adjusted to 8.0 with NaOH solution, and then the reacting temperature increased until 40°C. Subsequently, the 1st urea was added into a four-neck reactor to get initial F/U molar ratio of 2.0 and then reacting temperature and pH was adjusted to 90°C and 8.0, respectively, and then kept for 1 hour. After the alkaline reaction, the temperature and pH decreased to 80°C and 4.6 with formic acid solution for the condensation. This acidic condensation was stopped when the target viscosity reached the “J-K” scale based on a bubble viscometer (VG 900, Gardener-Holdt, Columbia, South Carolina, USA). Subsequently, the pH was adjusted to 8.0 with sodium hydroxide prior to the addition of the 2nd urea to obtain the final F/U molar ratio of 1.0, and then the temperature was kept at 60°C for 20 min. After that, the resins were cooled to room temperature and the pH was adjusted to 8.0.
For the in situ modification, 0.5 wt% of CNMs based on the theoretical non-volatile solid content was added to the reactor just prior to the 2nd urea addition at the pH of 8.0. Then, the CNMs were mixed in the resins using homogenizer for 10 min. Then the 2nd urea was added to the reactor to keep the reaction at 60°C for 20 min. The remaining steps was the same as the neat resins. For the post-mixing modification, CNMs were directly mixed with the synthesized resins using homogenizer for 10 min.
The properties of neat and modified UF resins such as non-volatile solid contents, viscosity, and gel time were measured to characterize these resins. The non- volatile solid contents of each UF resin were determined by measuring the mass before and after drying 1 g of the sample on aluminum dishes in an oven at 105°C. The viscosity of each UF resin was measured with a viscometer (DV-Ⅱ+, Brookfield, Middleboro, MA, USA) with spindle No. 82 at 20 rpm. The gel time was measured at 100°C by a gel time meter (Davis Inotek Instrument, North Carolina, USA). Each sample of UF resin was added to glass tubes with 3 % NH4Cl (based on the non-volatile solid contents) for measuring the gel time.
A X-ray diffractometer (D/Max-2500, Rigaku MiniFlex Ⅱ, Japan), with a Cu Kα radiation source (λ = 0.154 nm) was used for the calculation of the crystallinity and crystallite size of neat and modified UF resins. The scan range was from 2o to 50o, with a 0.02o/min step size. Each sample was prepared by mixing 3 % NH4Cl (based on the non-volatile solid content) and cured at 120 ℃ for 1 h.
The crystallinity of the samples was calculated from the XRD patterns with the peak deconvolution method with a Gaussian function (Wibowo and Park, 2020b) by using the OriginPro 9.0 software (OriginLab Corporation, Northampton, MA, USA). The equation for calculation the crystallinity of each sample is as follows:
where, Sc represents the area of the crystalline domain and St represents the total area (crystalline and amorphous). The crystallite size was calculated using the Scherrer equation (Park and Causin, 2013), as shown:
where, k is a constant factor (k = 1), λ is the wavelength of the X-ray radiation (λ = 0.154 nm), β is the width at half-maximum intensity in radians (FWHM), and θ is Bragg’s angle in radians. The peaks at 2θ= 21.7o, 24.4 o, 31.4 o, 40.2o were chosen for the calculation.
3. RESULTS and DISCUSSION
ATR-IR spectroscopy was utilized to characterize the chemical groups of CMFs, CNFs, and TEMPO- CNFs. All spectra of the CNMs are shown in Fig. 1 and detailed band assignments about each band are given in Table 1.
| Wavenumber (cm-1) | Group | Ref. |
|---|---|---|
| 3327-3330 | O-H stretching (hydrogen bond) | Chen et al., 2016; Fan et al., 2012; Pandey, 1999 |
| 2893-2898 | C-H stretching | Chen et al., 2016; Fan et al., 2012; Pandey, 1999 |
| 1632-1639 | O-H bending of absorbed water, conjugated C=O | Chen et al., 2016; Pandey, 1999 |
| 1600 | Asymmetric stretching of the carboxylate groups and O-H bedning of the absorbed water | Qu et al., 2021 |
| 1428 | CH2 symmetric bending | Chen et al., 2016; Fan et al., 2012 |
| 1367-1368 | C-H deformation | Chen et al., 2016; Pandey, 1999 |
| 1333-1334 | O-H in-plane deforamtion | Pandey, 1999 |
| 1314-1316 | CH2 bending | Chen et al., 2016; Fan et al., 2012; Pandey, 1999 |
| 1203 | O-H deformation | Pandey, 1999 |
| 1157-1160 | C-O-C asymmeric stretching | Chen et al., 2016; Pandey, 1999 |
| 1102-1105 | β-glycosidic ether linkages (C-O-C) from wagging, deformation and twisting of anhydro-glucopyranose ring | Chen et al., 2016 |
| 1050-1051 | C-O-C pyranose ring skeleton | Chen et al., 2016 |
| 1020-1028 | C-O stretch | Pandey, 1999 |
| 895-897 | Glucose ring stretch,C1-H deformation | Pandey, 1999 |

All CNMs have almost similar absorption bands. The absorption bands at 3327-3330 cm-1, 2893-2898 cm-1, 1428 cm-1, 1367-1368 cm-1, 1157-1160 cm-1, 1050-1051 cm-1, and 895-897 cm-1 are typical peaks of cellulose (Kondo and Sawatari, 1996). Kose et al., (2011) reported that the surface area of nanocellulose treated with aqueous counter collision (ACC) increased with an increase in the number of treatments. The hydroxyl groups also increased as the number of treatments increased. In spite of similarity of the absorption bands, there was a new absorption band at 1600 cm-1 in TEMPO-CNFs. This band was assigned to carboxylate groups of TEMPO-CNFs. Because TEMPO-CNFs are cellulose nanofibrils which have carboxylate groups at C6 on each cellulose nanofibril surface produced by 2,2,6,6-tetramethylpiperidine-1- oxylradical (TEMPO)/NaBr/NaClO oxidation at pH 10-11 or TEMPO/NaClO/NaClO2 oxidation at pH 4.6-6.8 (Isogai et al., 2011), this band clearly shows the presence of the carboxylate groups of TEMPO-CNFs. As the hydroxyl groups at C6 are substituted by carboxylate groups, the hydroxyl groups of TEMPO-CNFs decreases than those of CMFs and CNFs.
TEM analysis was conducted to show the morphological characteristics of CNMs. Typical TEM images were shown in Fig. 2 and the width of each sample was presented in Table. 2.
| Samples | Width (nm) |
|---|---|
| CMFs | 101 ± 69 |
| CNFs | 48 ± 41 |
| TEMPO-CNFs | 33 ± 14 |
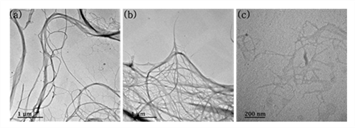
Both CMFs and CNFs had a similar morphology because they were produced by the same method of aqueous counter collision (ACC). This morphology was also reported in another paper (Kondo et al., 2014). A main difference between CMFs and CNFs was the width and this difference came from the number of collisions in the ACC method. Kondo et al., (2014) reported that the width of cellulose treated with ACC was decreased with increasing of ACC treatments. As displayed in Fig. 2 (c), TEMPO-CNFs showed their fibrillar morphology, and this had been reported by many researchers (Isogai et al., 2011; Saito et al., 2007; Saito and Isogai, 2004).
The properties of modified UF resins with different modification methods were shown in Table 3. The addition of CNMs by in situ and post-mixing method decreased the non-volatile solid contents, but increased the viscosity and gel time of the modified UF resins. A decrease of non-volatile solid contents was due to the extra water in TEMPO-CNFs (Moslemi et al., 2020; Veigel et al., 2012), because TEMPO-CNFs were added as suspension state. The extra water also decreased the viscosity as reported by Veigel et al., (2012). There were two reasons for an increase in the gel time. One was lower non-volatile solid contents (Moslemi et al., 2020; Veigel et al., 2012). The lower non-volatile solid contents could cause a decrease of the reactants in UF resins, which increased the gel time. The other one was a decrease of acidity due to extra water of TEMPO-CNFs (Moslemi et al., 2020). For curing, acidic condition is necessary, but extra water could decrease the acidity, and subsequently increased the gel time. When in situ and post-mixing were compared, there was no difference in the non-volatile solid contents, but the viscosity and gel time were different. However, the non-volatile solid contents of the modified UF resins was still lower than that of neat UF resins. Both the viscosity and gel time were higher than that of neat UF resins.
The properties of neat and modified UF resins were shown in Table 4, and each of these UF resins modified with in situ method. As mentioned above, the non-volatile solid contents decreased while the viscosity and gel time increased for the modified UF resins with TEMPO-CNFs. However, the non-volatile solid contents and viscosity increased while the gel time decreased for the modified UF resins with CMFs. This result maybe related to the state of CMFs. The UF resins were modified with wet CMFs after their filtration to reduce effects of moisture. As a result, the solid contents increased with the addition of CMFs while the gel time decreased because the reactant increased and acidic condition of the modified UF resins was not effected. For the UF resins modified with CNFs, there was a problem of the aggregation which caused by hydrogen bonds between fibrils (Chirayil et al., 2014). Like CMFs, CNFs were also filtered, but CNFs were aggregated more than CMFs, and did not disperse well in the UF resins. So, we added extra water for dispersing CNFs in the UF resins, which decreased the properties of the modified resins.
XRD patterns of the modified UF resins by two methods were not different from that of neat UF resins no matter which CNMs were used. XRD analysis was conducted to calculate the crystallinity and crystallite size of the modified UF resins. Fig. 3 shows XRD patterns, calculated crystallinity, and crystallite size of the modified UF resins by in situ and post-mixing. Fig. 4 also shows XRD patterns, calculated crystallinity, and crystallite size of modified UF resins with in situ modification using CNMs. The XRD patterns of the modified UF resinis by two methods were not different from that of neat UF resins no matter which CNMs were used. They still had four crystalline peaks as reported (Lubis and Park, 2020; Park and Causin, 2013; Park and Jeong, 2011; Wibowo and Park, 2020b).
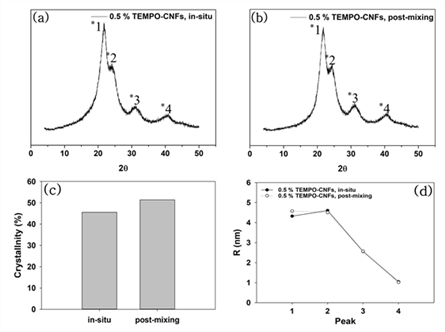
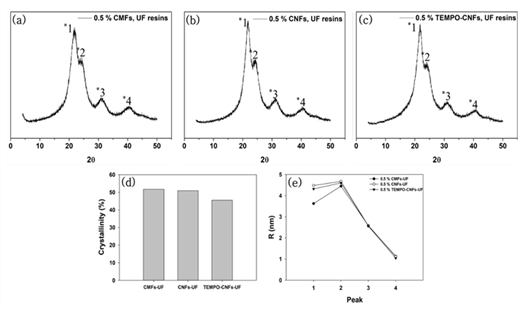
This result indicates that there is no change in the crystalline structure of the modified UF resins. There could be three reasons for this result. One of the reasons is an inability of CNMs to block the hydrogen bonds between the linear molecules in LMR UF resins. It is necessary to prevent the hydrogen bonds between linear molecules in LMR UF resins to convert crystalline structure (Wibowo and Park, 2020a; Wibowo et al., 2020a, 2020b). Therefore, in order to convert the crystalline structure of LMR UF resins to amorphous one, CNMs need to be modified to have highly reactive functional groups, which could prevent the hydrogen bonds between linear molecules in LMR UF resins. Other reason is the size of CNMs. Because a size of the hydrogen bonds between linear molecules in the LMR UF resins was at the molecular level, it may have been difficult for CNMs to prevent the hydrogen bonds. In a previous report (Wibowo et al., 2020a), modified nanoclay changed the crystallinity of LMR UF resins by intercalation, as nanoclay had nanoscale in all sizes. However, it is difficult for CNMs to react with linear molecules in LMR UF resins as the modified nanoclay because CNMs have at least one nanoscale dimension and entangled structure (Nechyporchuk et al., 2016; Phanthong et al., 2018). In other words, the CNMs that have poorly accessible hydroxyl groups and much bigger size are not possibly prevented the formation of hydrogen bonds because the formation of hydrogen bonds between linear molecules requires molecular level interaction between them.
Another reason maybe related to the low addition levels of CNMs. To modify the crystalline structure of LMR UF resins to amorphous one, 5 % modified nanoclay (Wibowo et al., 2020a), and 20 % melamine (Wibowo and Park, 2020a) were needed. However, in this research, only 0.5 % CNMs was used for modification due to the agglomeration of CNMs. So, it is necessary to develop a method of increasing the addition level of CNMs without agglomeration. When the crystallinity was compared between in situ and post-mixing, the in situ method had lower crystallinity than that of post-mixing, but there was no difference in crystallite size between two methods. The lower crystallinity of the in situ method may be resulted from the interaction of CNMs with reacting substance. CNMs could be reacted with three kinds of substances such as condensated molecules, second urea, and free-formaldehyde in the modified UF resins. But, unlike the in situ modification, CNMs in the post-mixing could be difficult to react with condensated molecules or second urea because they were added after the reactions. Contrary to the crystallinity, there was not much difference in the crystallite size between in situ and post-mixing method. Although the CNMs didn’t convert the crystalline domain to amorphous one, the in situ modification was more effective in decreasing the crystallinity of LMR UF resins. When the crystallinity among the CNMs was compared, modified UF resins with TEMPO-CNFs had the lowest crystallinity. Since TEMPO-CNFs had carboxylate groups, it could form an ester with hydroxylmethylated urea (Fischer and Speier, 1895). In addition, acidic media facilitated the esterification between TEMPO-CNFs and OH groups on hydroxylmehylated ureas because the OH groups on hydroxylmethylated ureas could be easily reacted at acidic condition (Akinterinwa et al., 2020). Therefore, the modified UF resins with TEMPO-CNFs had the lowest crystallinity among the CNMs. Furthermore, When all CNMs were compared, the crystallite sizes had difference for the main peak of XRD patterns (Park and Causin, 2013; Wibowo et al., 2020b). A decrease of this peak meant that the modified UF resins with CMFs had larger domains and less closely spaced (Park and Causin, 2013). Because CMFs had greater width than those of CNFs and TEMPO-CNFs, the greater width maybe contributed to the decrease of crystallite size.
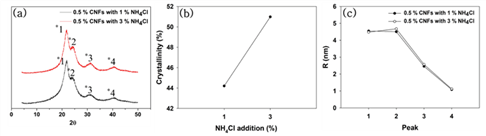
Fig. 5 shows the effect of hardener addition on the XRD patterns, crystallinity, and crystallite size of the modified UF resins by the in situ method. There was no difference in the XRD pattern and crystallite sizes, but there was a little difference in the crystallinity. The higher addition level of hardener was, the higher crystallinity was. This phenomenon already reported by other papers. Park and Jeong (2011) reported that addition level of hardener was not related to the crystalline peak position but intensity and extent of crystalline regions. Furthermore, it was inevitable for higher addition levels of hardener to increase of crystallinity due to higher level of addition accelerated the catalyzing effect and subsequently formed tight linear structure in LMR UF resins (Wibowo and Park, 2020b).
4. CONCLUSION
In this study, the LMR UF resins were modified by two different methods (in situ and post-mixing) using three different CNMs (CMFs, CNFs, and TEMPO- CNFs). It was confirmed that TEMPO-CNFs had carboxylate groups and conserved their fibrillar morphology, and that CMFs had the greatest width. The viscosity and gel time of the in situ and post-mixing method with TEMPO-CNFs decreased the non-volatile solid contents while increased the viscosity and gel time. In addition, the modified UF resins with the in situ method using CMFs increased the non-volatile solid contents and viscosity, but decreased the gel time. Both in situ and post-mixing did not change the crystalline domain to amorphous one, and the crystallite size was not also changed. But, the in situ modification had a lower crystallinity than that of the post-mixing. The addition of all CNMs by the in situ modification did not change the crystalline domain of the modified UF resins to amorphous one, but the crystallinity of UF resins modified with TEMPO-CNFs was the lowest. Furthermore, the crystallite sizes of UF resins modified with CMFs was the lowest. Besides, the addition level of hardener affected to the crystallinity, and a higher addition levels lead to a higher crystallinity. In addition, it is suggested that the modification of LMR UF resins using CNMs requires to develop a method for avoiding agglomeration of CNMs in UF resins, and also to modify the CNMs’ hydroxyl groups to reactive functional groups to prevent the hydrogen bonds between linear molecules of LMR UF resins.