






1. INTRODUCTION
Terpenoids are secondary plant metabolites with important protective roles and a variety of applications in various fields such as fragrances and flavors, pharmaceutical agents, and also as insecticides (Tholl, 2006; Bicas et al., 2008).
Terpenes are biosynthesized from the C5 universal precursors isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP). Prenyltransferases assemble IPP and DMAPP into linear prenyl diphosphate precursors such as farnesyl diphosphate (FPP, C15), which are then rearranged by terpene synthases (TPS) into a variety of distinct terpenes such as sesquiterpenes (C15) (Peralta-Yahya et al., 2011; Schnee et al., 2002). Sesquiterpenes, composed of three isoprene units, exhibit diverse antioxidant, antimicrobial, and antitumor bioactivities (Sandler et al., 2006). Although sesquiterpene compounds are naturally produced in plants, their yields from biological sources is generally low. In addition, the chemical synthesis of natural products is a difficult and expensive procedure (Huang et al., 2008). Owing to the availability of metabolic engineering systems and fermentation techniques, microorganisms have been established as beneficial platforms for the mass production of useful substances, and as alternatives to less productive plants. Various recombinant fungal enzymes have so far been produced by engineered microbial platforms and are directly used for the biobleaching of softwood kraft pulp (Kim et al., 2007).
Previously, we found that Polyporus brumalis, white rot fungus, has a unique ability of synthesizing eudesmane-type sesquiterpenoids as secondary metabolites (Lee et al., 2016). Furthermore, TPS, which is highly expressed in the presence of magnesium by P. brumalis, identified by a transcriptome analysis and sequencing of the full-length terpene synthase, including a metal-binding motif (DEXXD) (Gao et al., 2012), has been isolated by RACE (rapid amplification of cDNA ends)-PCR. TPS of P. brumalis is phylogenetically closely related to TPS of other basidiomycetes. The amino acid similarity between this sequence compared with that of Neolentinus lepideus HHB14362 ss-1 is 69%, Gloeophyllum trabeum ATCC 11539 is 68%, and Fomitiporia mediterranea MF3/22 is 65% (Lee et al., 2017). TPS is assumed to be necessary for the formation of a cyclic structure by removing a phosphate group from FPP and inducing cyclization in sesquiterpenoids (Lee et al., 2017). Therefore, we tried to transformation the P. brumalis terpene synthase gene in Pichia pastoris to produce a recombinant enzyme and functionally characterized this recombinant TPS.
2. MATERIALS and METHODS
P. brumalis was obtained from the Korean Collection for Type Cultures (KCTC46459). Fungi were inoculated onto potato dextrose agar (BD Difco, Franklin Lakes, NJ, USA) and were incubated in the stationary phase at 28°C for 7 days. Subsequently, mycelia were fully grown on Petri dishes and a blade was used to separate the mycelial layer covering the agar medium from the medium. Then, the mycelium were homogenized for 30 s in 20 ml of distilled water and the homogenized suspension was inoculated into a modified medium including magnesium (Lee et al., 2016). The inoculated fungus was incubated for 7 days at 28°C under 120 rpm shaking.
Total RNA was extracted with Hybrid-R (GeneAll, Seoul, Korea) from the incubated P. brumalis mycelium (described above). The 2100 Bioanalyzer (Agilent, Santa Clara, CA, USA) was used to determine the amount and quality of RNA. cDNAs were synthesized by transcribing 1 μg of RNA with MMLV reverse transcriptase (Promega, Madison, WI, USA), as recommended by the manufacturer. Fragments, approximately 1.2 kb in size, were amplified with pairs of TPS ORF primers (Forward: 5′-TGTACTATGCT CACTTCCACCA-3′ and Reverse: 5′-CTCATCCC ACGGTTACAGG-3′). PCR was performed as follows: initial denaturation at 94°C for 3 min, 30 cycles of denaturation at 94°C for 1 min, annealing at 55°C for 1 min, extension at 72°C for 3 min, and a final 5-min extension at 72°C. The amplified fragments were sub-cloned into a pCR2.1-TOPO (Invitrogen, Carlsbad, CA, USA) vector and the plasmid DNA was sequenced.
The terpene synthase open reading frame (ORF) was separated from pCR2.1-TOPO by digesting with EcoRI and NotI restriction enzymes and was inserted into the pPICZαC vector (Invitrogen) linearized with EcoRI and NotI. The combined cassette was amplified in the Escherichia coli DH5α strain using heat shock (Sambrook and Russell, 2001) and the selection was performed on low-salt LB agar medium (BD Difco) containing 25 μg/ml zeocin.
P. pastoris X33 was transformed by electric shock (following methods described in the Pichia expression system manual, with slight modifications; Invitrogen). Plasmid DNA was digested with PmeI restriction enzyme and was linearized prior to transformation. For pre-culture, a single P. pastoris colony was inoculated in 5 ml of YPD medium (1% Bacto-Yeast Extract, 2% Bacto-Peptone, 2% dextrose) and incubated overnight at 30°C under 180 rpm shaking. The incubated culture was re-inoculated in fresh YPD medium (180 rpm, 30°C) and was allowed to grow until an OD600 of 1.3-1.5 was reached. Then, the cells were harvested by centrifugation at 4000 rpm for 5 min at 4°C and the pellet was washed twice with 20 ml of ice-cold sterile water. A final round of washing was performed with ice-cold 1 M sorbitol and the cell pellet was resuspended in 0.4 ml of ice-cold 1 M sorbitol. An aliquot of yeast cells (80 μl) was mixed with 0.1 μg of linearized plasmid DNA. This mixture was transferred to a chilled electroporation cuvette (0.2 cm gap) and was subjected to a pulsed at 2 kV, 25 μF, 200 Ω (Bio-Rad Gene Pulser; Hercules, CA, USA). The cell suspension with 1 M sorbitol was transferred to a 50 ml centrifuge tube and was incubated at 30°C under 180 rpm shaking. After incubation for 1 h, the cells were spread on YPD plates supplemented with zeocin (100 μg/ml) and were incubated at 30°C until colonies appeared. All the transformants were confirmed by colony PCR and sequencing.
Single transformant colonies were selected from agar plates and were transferred to 5 ml of BMGY medium (1.34% yeast nitrogen base with ammonium sulfate without amino acids, 1% yeast extract, 2% peptone, 100 mM potassium phosphate (pH 6), 0.4 μg/ml biotin, 10% glycerol) in a 50 ml Falcon tubes and incubated at 30°C and 200 rpm for 18 h. Subsequently, 0.5 ml of this seed culture was inoculated into 50 ml of BMGY medium in a 250 ml flask and was grown until an OD600 of 4-6 was reached. The cultured cells were harvested by centrifugation at 4000 rpm for 5 min at 4°C and the pellet was washed with sterile water.
The pellet was resuspended in 50 ml of BMMY medium (1.34% yeast nitrogen base with ammonium sulfate without amino acids, 1% yeast extract, 2% peptone, 100 mM potassium phosphate (pH 6), 0.4 μg/ml biotin, 0.5% methanol) and incubated for 72 h at 30°C and 200 rpm. Furthermore, to maintain a final concentration of 0.5% necessary for inducing the terpene synthase expression, methanol (100%) was added to 50 ml of BMMY medium every day.
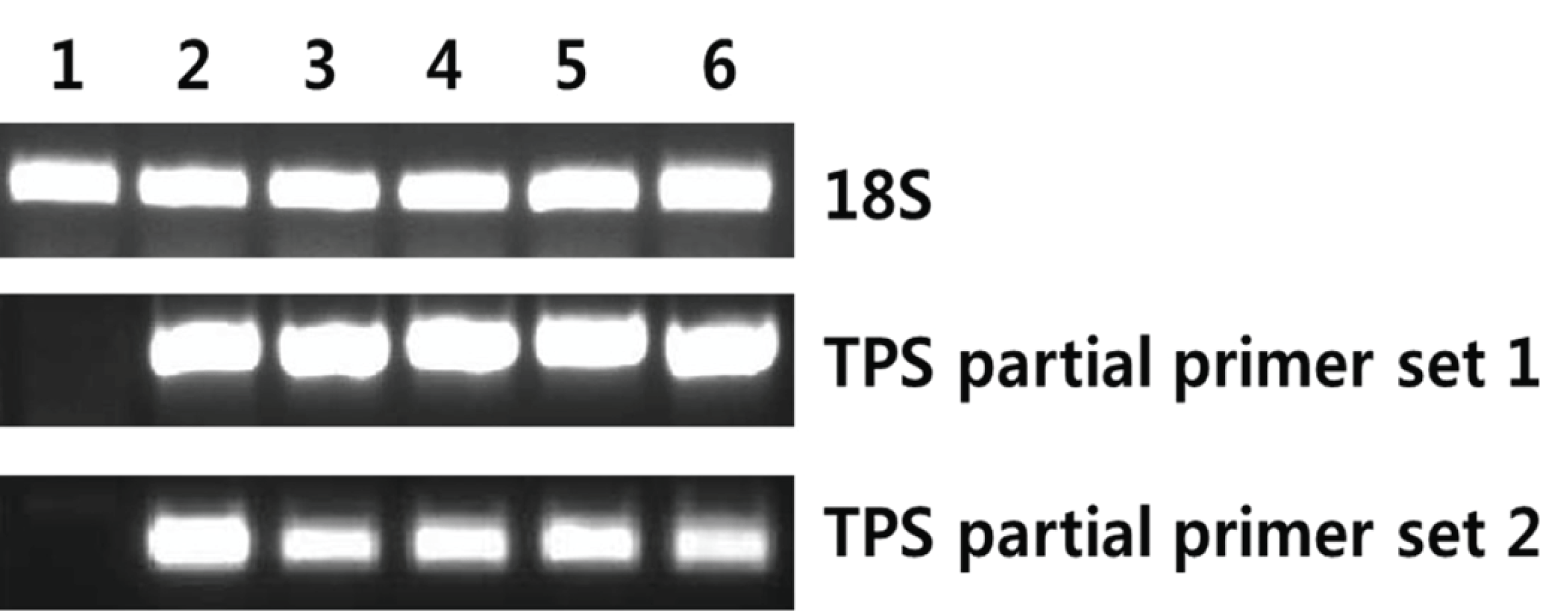
AccuPower RT/PCR Premix (Bioneer, Daejeon, Korea) was used to detect the mRNA encoding terpene synthase; 18S rRNA served as the internal control. The reaction mixture (20 μl) was analyzed by gel electrophoresis. Gene-specific PCR primers were designed from tail region of the TPS gene. TPS partial primer set 1 (F: 5′-ACCATTCGCACCATCGACAGC TACC-3′, R: 5′-GCTCATCCCACGGTTACAGGGCTC- 3′) generated a 630 bp product and TPS partial primer set 2 (F: 5′-GAACTCCGTGATTGTCCAGGC-3′, R: 5′-TACACCGCGACCTGCTTGT-3′) amplified a 414 bp product (Fig. 1). The expression level of 18S rRNA was determined by RT-PCR using commercial primers (QuantumRNA Universal 18S; Ambion, Foster City, CA, USA) to validate the concentration of cDNA.

The incubated cells (2.5.1) were harvested by centrifugation at 13,000 rpm for 1 min and resuspended in lysis buffer (100 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.5% Triton X-100, 1× protease inhibitor (leupeptin hemisulfate salt; Sigma, St. Louis, MO, USA)). The resuspended cells were disrupted by bead beating and debris were removed by centrifugation at 13,000 rpm for 15 min at 4°C. The proteins in the supernatant were resolved by 12% SDS-PAGE (Kim et al., 2013).
3. RESULTS and DISCUSSION
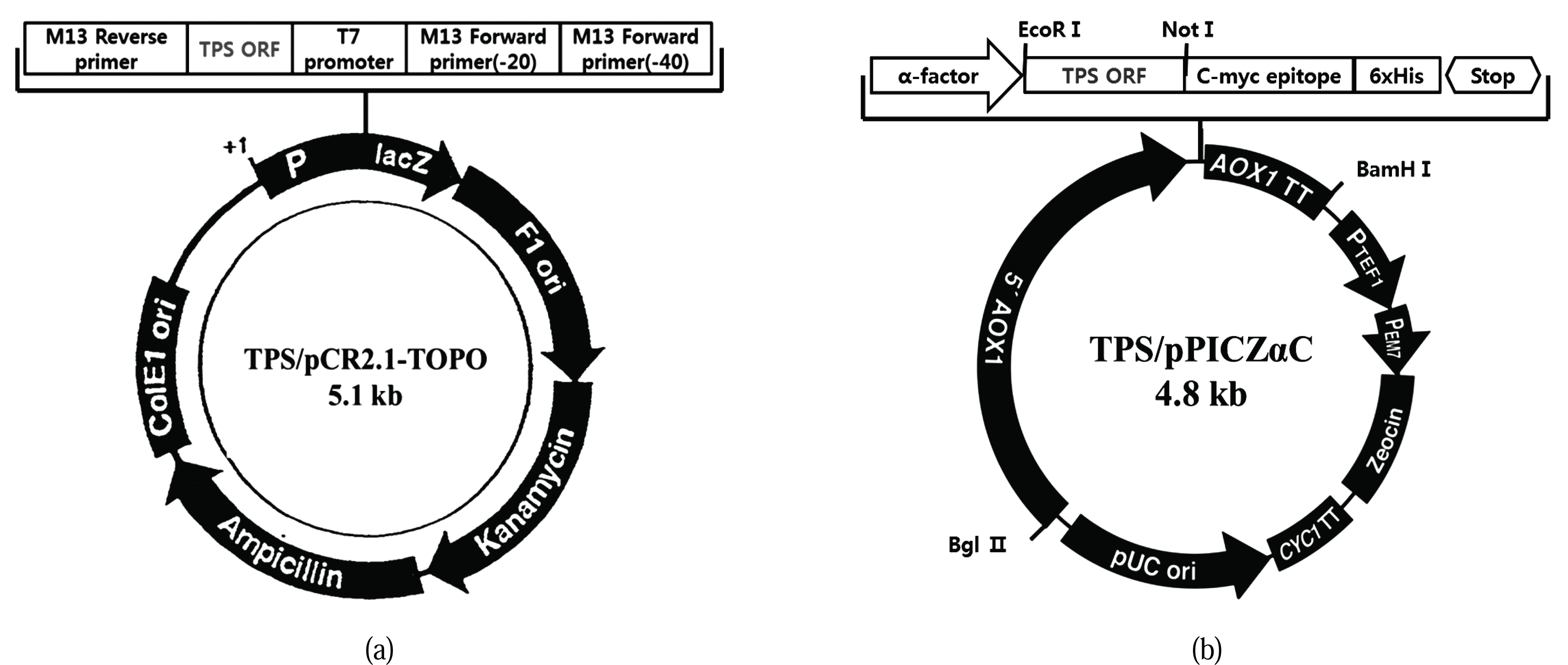
Terpene synthase genes have a metal-binding region (DEXXD, NSE) that is highly conserved among different taxa. In a previous study, we used RACE-PCR to identify the sequence of an unknown region flanked by conserved sequences and thereby obtained ORF sequences of approximately 1.2 kb size. For ease of insertion into the yeast expression vector, the terpene synthase gene ORF obtained by RT-PCR was subcloned into a pCR2.1-Topo vector and a TPS/pCR2.1- TOPO of approximately 5.1 kb was obtained (Fig. 2A). Following this, the TPS ORF of the pCR2.1-Topo clone was separated and was transferred to a Pichia expression vector, pPICZαC, in-frame with the α-factor signal sequence for secretion. This resulted in a 4.8 kb expression cassette, TPS/pPICZαC (Fig. 2B). The cassette for expression was analyzed by digestion and sequencing. These results indicated that all the cloning steps were performed correctly. EcoR1 and Not1 restriction enzymes were used to confirm the cloning. The structure contained a TPS ORF of approximately 1.2 kb (Fig. 3).
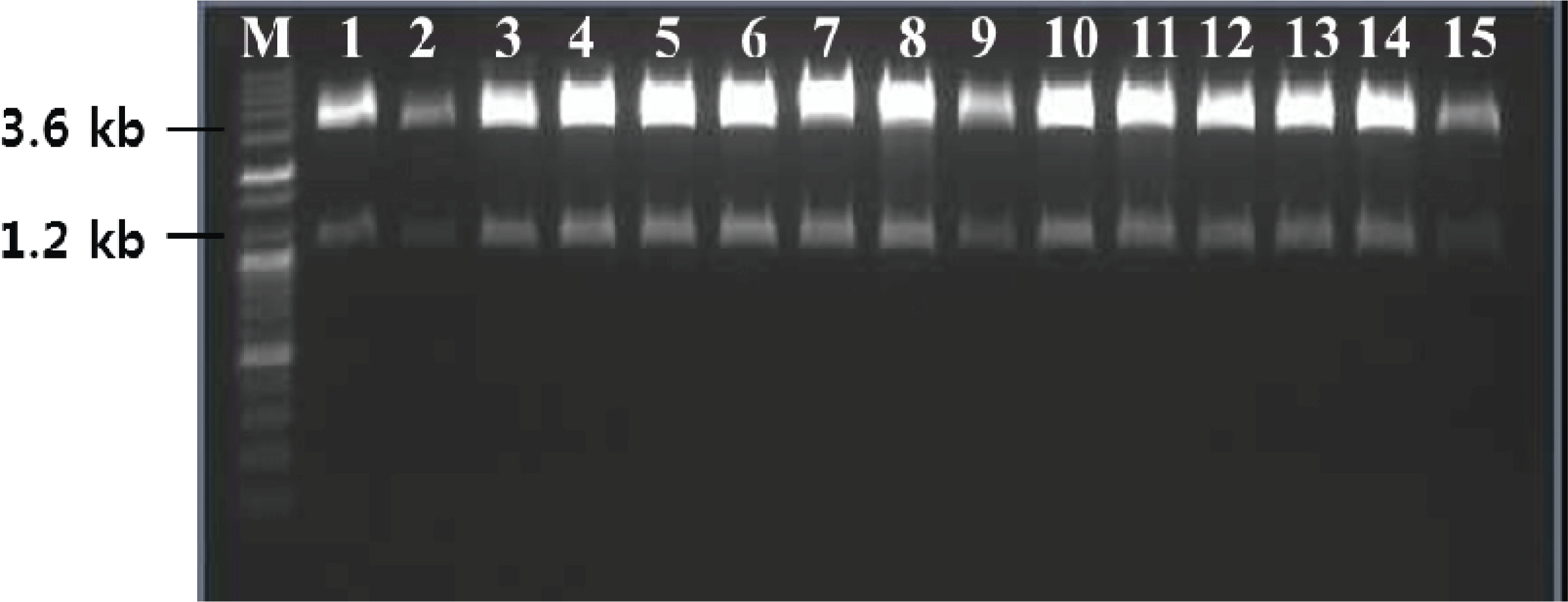
As a basis for functional and structural studies, the TPS cDNA was heterologously expressed in P. pastoris. The linearized DNA was directly introduced without carrier DNA and the transformed cells, obtained by using an electroporation device, were plated on YPD medium supplemented with zeocin (100 μg/ml). Out of the more than 20 obtained candidates, five healthy appearing candidate single cells were selected. The small number of transformants can be explained by the scale of the culture, the amount of plasmid used, transformation without carrier DNA, and dilution during the plating step. The estimated transformation efficiency was 8 × 102/1 μg of linearized DNA. Gene insertion in the resulting transformants was confirmed by colony PCR, which confirmed the size of the TPS ORF to be 1.2 kb (Fig. 4), and insertion of the gene without any mutations was confirmed by sequencing (not shown).
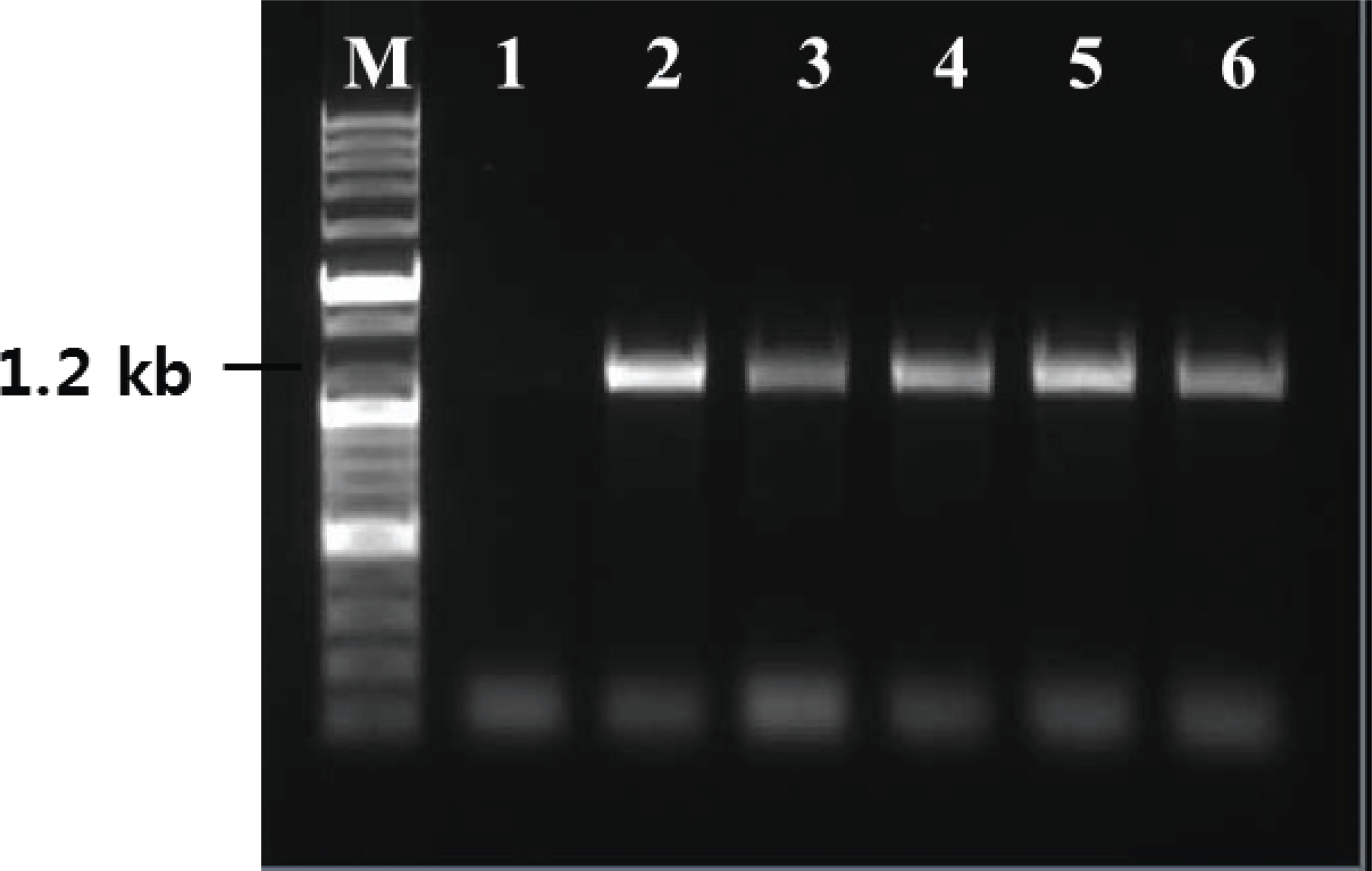
Yeast cells grown in BMGY medium, until an optical density of 2-6 at 600 nm, were transferred to an induction medium and were cultured for 72 h. RNA was extracted from the cultured yeast cells and was evaluated by RT-PCR using primers, which amplify the partial region of TPS ORF, to confirm expression. For all transformants, PCR products were obtained using TPS primers. However, wild-type yeast used as a negative control showed no amplification by RT-PCR using TPS targeting primers (Fig. 5). Thus, the TPS gene was properly expressed in the transgenic system.
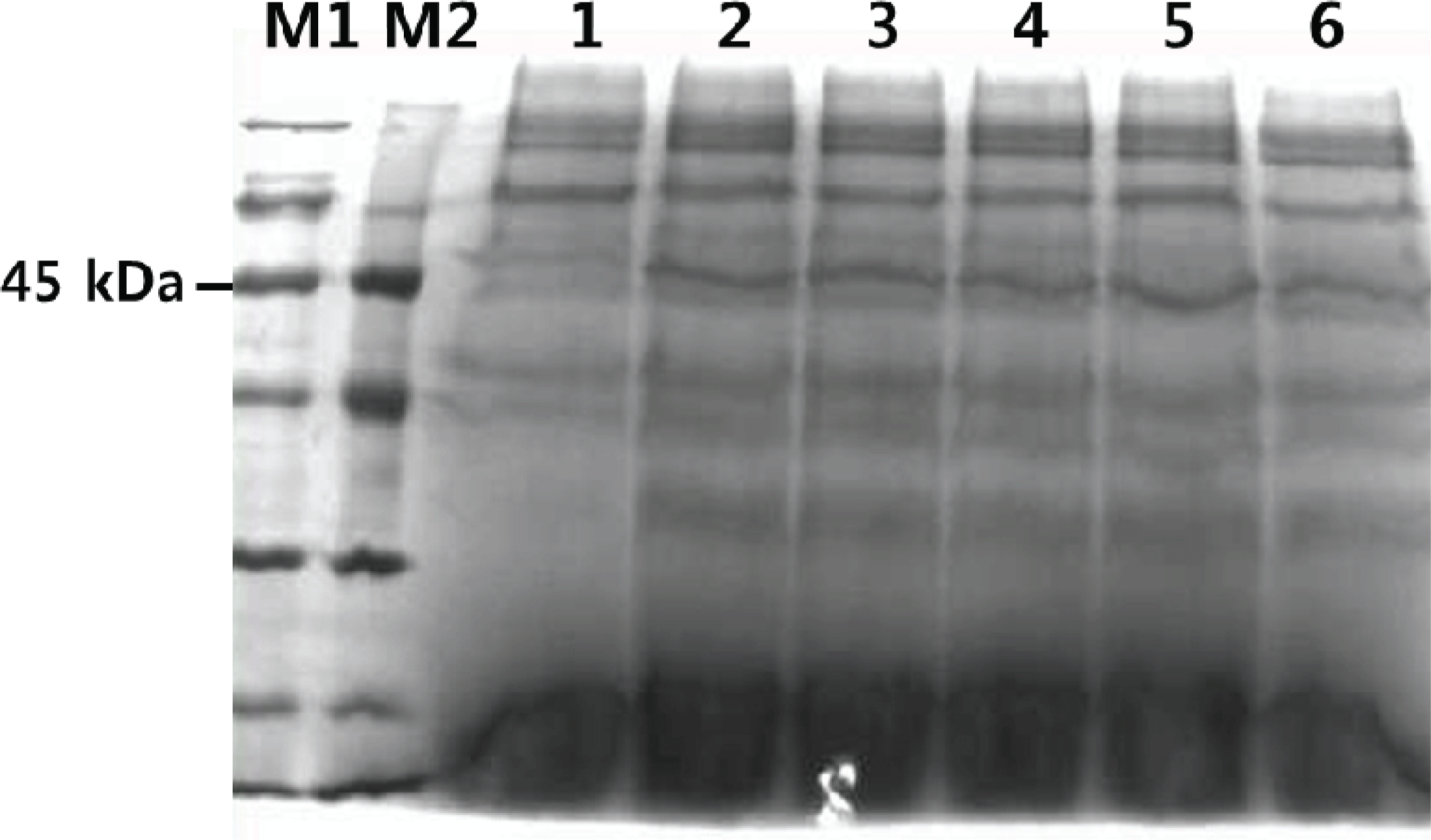
SDS-PAGE was used to confirm the expression of TPS in the transformants. As Fig. 6 shows, the molecular weight of the secreted TPS was about 45 kDa. This was consistent with the predicted molecular weight computed using Compute pI/Mw tool. The following factors may have contributed to the faint protein band: gene copy number, site and mode of chromosomal integration of the expression cassette, 5′ and 3′ untranslated regions of mRNA, A+T composition of cDNA, transcription and translation blocks, nature of the secretion signal, endogenous protease activity, host strain physiology, media and growth conditions, and fermentation parameters (Dong et al., 2006).
To evaluate the activity of the recombinant enzyme, we employed a colorimetric assay using malachite green pyrophosphate (MG). In this assay, a monophosphate (Pi) produced by the reaction of the substrate FPP with terpene synthase reacts with malachite green and results in a green color (Vardakou et al., 2014). MG assay was performed using culture media of each strain. Disappointingly, the results for both the wild type and the transformants were unclear (data not shown). Since the MG assay does not directly detect TPS activity, the components of the medium or other factors may have affected the results of this assay. A method for the direct measurement of TPS activity has not yet been established and should be a focus of future research.
P. pastoris was used in this study because it can be grown to high cell densities and genetic manipulation in this species is easier than in fungi. Other benefits of the P. pastoris system include the strong AOX1 promoters available to drive the expression of foreign proteins of interest (Cereghino et al., 2000; Ahmad et al., 2014), thus, enabling the production of large quantities of the target protein with relative technical ease and at a lower cost compared with most other eukaryotic systems (Daly et al., 2005; Jonsson et al., 1997). P. pastoris is a suitable host for eukaryotic heterologous expression systems and this system has been used to achieve a heterologous expression of the MnP5 gene (Lee et al., 2005). In this study, we obtained stable transgenic strains for the production of recombinant TPS using a heterologous expression. Further studies including the development of a method for measuring the activity of recombinant proteins generated by transformants are needed.
4. CONCLUSION
P. pastoris can be genetically manipulated to produce various compounds that are only produced naturally in higher eukaryotes. In this study, five TPS expressing transformants were obtained. We could confirm that genes of transgenic strains were expressed at transcription level. However, the activity of the recombinant proteins could not be confirmed. Thus, the establishment of a method for measuring TPS activity is necessary. The recombinant enzymes created in this study would prove useful in developing methods for measuring enzyme activity and in understanding the formation of the cyclic structure from FPP during sesquiterpenoid synthesis.